 Back
BackPrions and Intrinsically Disordered Proteins: Structure, Function, and Disease
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Prions: Protein Folding Gone Wrong
Definition and Discovery
Prions are proteinaecous infectious particles that lack nucleic acids but can transmit disease from cell to cell. Discovered and named in 1982, prions represent a unique class of infectious agents, with their pathogenicity arising from protein misfolding rather than genetic material.
Prion = Protein + Infection
Associated with neurodegenerative diseases such as Scrapie (sheep), Kuru and Creutzfeldt-Jakob Disease (CJD) (humans).
Other diseases with prion-like mechanisms: Huntington's Disease (HD), Parkinson's Disease (PD), Amyotrophic Lateral Sclerosis (ALS), Type 2 Diabetes.
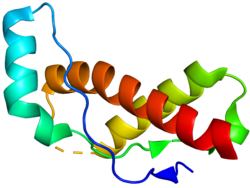
Structural Biology of Prions
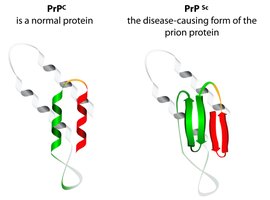
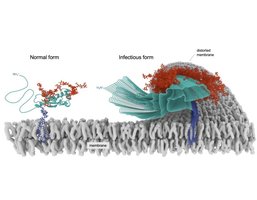
The prion protein exists in two main conformations: the normal cellular form (PrPc) and the pathogenic form (PrPsc). Both share the same amino acid sequence, but their structures and functions differ dramatically.
PrPc: Mostly α-helical, soluble, encoded by the PRNP gene, processed to ~210 amino acids.
PrPsc: Largely β-sheet, prone to aggregation, highly stable, forms amyloid fibrils.
Structure determines function: Misfolding leads to disease.
PrPc and Memory Function
Evidence suggests that PrPc plays a role in memory. Mice lacking PrPc function (PrP-) show deficits in long-term memory compared to wildtype mice. In Drosophila, PrP-like proteins localize to neuron synapses, possibly stabilizing neuronal connections and supporting memory.
Wildtype mice improve in maze navigation after training; PrP- mice do not.
PrP-like proteins may stabilize synaptic connections.
Normal vs Mutant Prion Protein
Mutations in the PRNP gene can lead to autosomal dominant inheritance of prion diseases such as CJD. The mutation alters the amino acid sequence, promoting misfolding and disease.
Prions are not inherited through the germline but can be transmitted horizontally (cell-to-cell).
Mutations are inherited vertically (from parent to offspring).
Prion Infectivity and Disease Transmission
PrPsc's misfolded structure is self-propagating, inducing normal PrPc to adopt the pathogenic conformation. Prion diseases can be transmitted horizontally, and mutations in PRNP are inherited vertically.
Example: Parkinson's Disease treatment with fetal cell implants led to prion invasion of implanted cells.
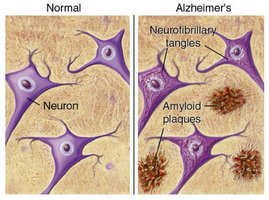
Alzheimer's Disease: Plaques and Tangles
Pathological Features
Alzheimer's Disease is characterized by the formation of amyloid-beta plaques and neurofibrillary tangles (Tau complexes). These features cause mitochondrial damage, cell death, and are classified as "Tauopathies" due to their prion-like behavior.
Plaques: Amyloid-beta complexes.
Tangles: Tau protein aggregates.
Spread via neuronal contacts; ALS and other diseases also classified as Tauopathies.
Formation of Fibrils and Amyloid Plaques
Prion Aggregation and Amyloid Formation
Prions are examples of intrinsically disordered proteins (IDPs) or contain intrinsically disordered regions (IDRs). These proteins continuously change conformation, and when two PrPsc molecules interact, they stabilize and form growing fibrils, eventually leading to amyloid plaques.
Not all amyloids are transmissible; prions are a subset of amyloids.
Prion disease can produce cardiac amyloidosis in mice.
Intrinsically Disordered Proteins (IDPs) and Regions (IDRs)
Structural Characteristics
IDPs and IDRs lack a stable three-dimensional structure under physiological conditions but are fully functional. This challenges the traditional structure-function paradigm in molecular biology.
Exist as dynamic ensembles of structures.
Enriched in polar and charged amino acids (e.g., Lys, Glu, Ser, Pro).
Depleted in hydrophobic residues that form stable cores.
Functional Roles of IDPs/IDRs
Despite lacking fixed structure, IDPs are biologically important due to their flexibility, which allows:
Molecular recognition: Become structured upon binding partners ("coupled folding and binding").
Signaling and regulation: Act as hubs in signaling pathways.
Scaffolding: Bring together different proteins or complexes.
Transcriptional regulation: Many transcription factors have large IDRs for DNA binding and partner interactions.
Ordered vs. Disordered Proteins: Summary Table
Property | Ordered Proteins | Disordered Proteins (IDPs/IDRs) |
|---|---|---|
Structure | Stable, well-defined 3D structure | Dynamic, ensemble of conformations |
Amino Acid Composition | Hydrophobic core, less polar/charged residues | Enriched in polar/charged residues, depleted hydrophobic |
Function | Enzyme activity, structural support | Signaling, regulation, molecular recognition |
Examples | Hemoglobin, actin | Prion protein, transcription factors |
Prion Disease Pathology
Spongiform Encephalopathies
Prion disease in the brain destroys neurons, creating vacuoles and "spongiform encephalopathies." These vacuoles represent regions of cell death and are a hallmark of prion-induced neurodegeneration.
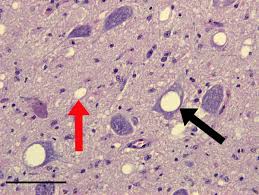
Formation of PrPsc Aggregates
PrPsc forms fibrils and amyloid plaques in brain cells, leading to progressive neurodegeneration.
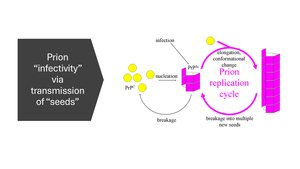
Prion Replication Cycle
Prion infectivity is mediated by the transmission of "seeds" that induce conformational changes in normal proteins, propagating the misfolded state.
Cycle involves nucleation, elongation, conformational change, and breakage into new seeds.
Summary
Prions are infectious proteins that cause disease through misfolding and aggregation.
Intrinsically disordered proteins (IDPs) play important roles in cellular regulation and signaling.
Prion diseases and tauopathies (e.g., Alzheimer's, ALS) share mechanisms of protein aggregation and cell death.
Additional info:
Some academic context was inferred regarding the molecular mechanisms of prion propagation and the functional roles of IDPs/IDRs.
Tables and diagrams were recreated and expanded for clarity and completeness.
