 Back
BackFree Energy, Nonstandard States, and Equilibrium in Chemical Reactions
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Free Energy Changes for Nonstandard States
From Standard Free Energy (ΔG°) to Actual Free Energy (ΔG)
In real chemical systems, conditions often differ from the standard state (1 M concentration, 1 atm pressure). The Gibbs free energy change under these nonstandard conditions is given by:
Key Equation:
R: Universal gas constant, 8.314 J/(mol·K)
T: Temperature in Kelvin
Q: Reaction quotient, reflecting current concentrations/pressures
Physical Interpretation:
When Q = 1 (standard conditions):
When Q < K: (forward reaction is spontaneous)
When Q > K: (reverse reaction is spontaneous)
When Q = K: (system at equilibrium)
The term corrects the standard free energy for the actual conditions of the system.
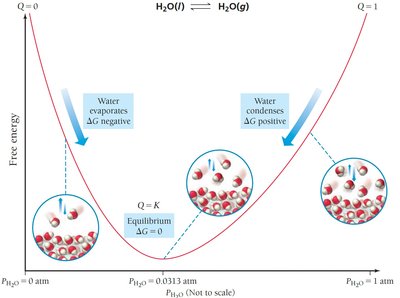
Free Energy and Equilibrium: Relating ΔG° to K
Derivation and Interpretation of ΔG° = −RT ln K
At equilibrium, the reaction quotient Q equals the equilibrium constant K, and the free energy change ΔG is zero. Substituting these values into the key equation yields:
Therefore,
Or, rearranged:
Implications:
Small changes in ΔG° cause large changes in K due to the logarithmic relationship.
ΔG° < 0: K > 1 (products favored at equilibrium)
ΔG° > 0: K < 1 (reactants favored at equilibrium)
ΔG° = 0: K = 1 (reactants and products equally favored)
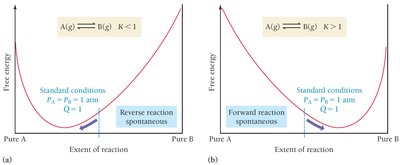
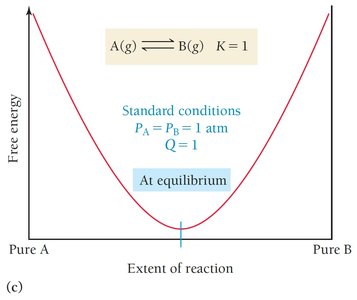
Summary Table: Relationship Between ΔG° and K
The following table summarizes how the sign and magnitude of ΔG° relate to the equilibrium constant K and the position of equilibrium:
ΔG°rxn | K value | Equilibrium Position | Reaction at Standard Conditions |
|---|---|---|---|
ΔG° << 0 (large negative) | K >> 1 | Products strongly favored | Spontaneous forward |
ΔG° < 0 (moderately negative) | K > 1 | Products favored | Spontaneous forward |
ΔG° = 0 | K = 1 | Reactants = products | At equilibrium |
ΔG° > 0 (moderately positive) | K < 1 | Reactants favored | Spontaneous reverse |
ΔG° >> 0 (large positive) | K << 1 | Reactants strongly favored | Spontaneous reverse |
Additional info: Small changes in ΔG° produce large changes in K due to the exponential relationship.
Example: Calculating K from ΔG°rxn
Consider the reaction: N2O4(g) ⇌ 2 NO2(g). Given the following standard free energies of formation:
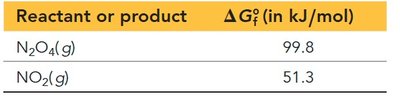
Calculate ΔG°rxn:
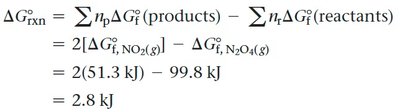
Calculate K at 298 K:
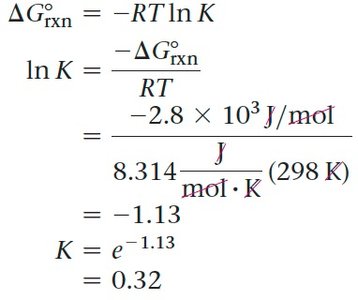
Result: K = 0.32, indicating that products are slightly disfavored at standard conditions.
Coupling Reactions: Driving Nonspontaneous Processes
Thermodynamic Coupling in Chemistry and Biology
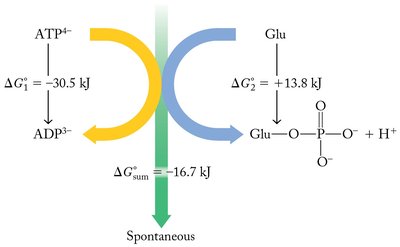
Some reactions with ΔG > 0 (nonspontaneous) can be made to occur by coupling them with a spontaneous reaction (ΔG < 0) so that the overall ΔG is negative. This principle is fundamental in both industrial and biological processes.
Industrial Example: Extraction of iron from iron ore (Fe2O3) is nonspontaneous, but becomes spontaneous when coupled with the combustion of carbon.
Biological Example: ATP hydrolysis (ΔG ≈ −30.5 kJ/mol) is coupled to many cellular reactions to make them spontaneous overall.
Key Equations in Thermodynamics and Equilibrium
Boltzmann Equation (Entropy):
Entropy Change (Reversible Process):
Entropy of Surroundings:
Gibbs Free Energy:
Standard Entropy Change:
Standard Free Energy Change:
Free Energy at Nonstandard Conditions:
Relationship to Equilibrium Constant:
Comprehensive Review: Final Takeaways
ΔG = ΔG° + RT ln Q: Corrects standard free energy for actual conditions.
ΔG° = −RT ln K: Master equation linking thermodynamics and equilibrium.
Large negative ΔG° → K ≫ 1 (products favored); large positive ΔG° → K ≪ 1 (reactants favored).
Coupling reactions: Nonspontaneous processes can be driven by coupling with spontaneous ones.
All spontaneous processes are driven by ΔS_universe > 0; ΔG = −TΔS_universe is the practical criterion.
Key equations: S = k ln W; ΔS = q/T; ΔS_surr = −ΔH/T; ΔG = ΔH−TΔS; ΔS°_rxn and ΔG°_rxn formulas; ΔG = ΔG° + RT ln Q; ΔG° = −RT ln K.
