 Back
BackDisorders of Haemostasis and Laboratory Investigation
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Disorders of Haemostasis
Overview of Haemostasis
Haemostasis is the physiological process that prevents excessive bleeding when blood vessels are injured. It involves a complex interplay between blood vessels, platelets, coagulation factors, and the fibrinolytic system. The process is divided into primary and secondary haemostasis, followed by fibrinolysis.
Primary haemostasis: Involves vasoconstriction, platelet adhesion, and aggregation to form a temporary platelet plug.
Secondary haemostasis: Activation of the coagulation cascade, resulting in the formation of a stable fibrin clot.
Fibrinolysis: Dissolution of the clot after vessel repair.
Role of Endothelium
The healthy endothelium expresses ecto-ADPase (CD39), prostacyclin (PGI2), and nitric oxide (NO), which inhibit platelet adhesion and activation. It also possesses anticoagulant mechanisms to prevent unnecessary clotting.
Platelet Disorders
Types of Platelet Disorders
Quantitative: Thrombocytopenia (low platelet count) or thrombocytosis (high platelet count).
Qualitative: Functional defects, which may be inherited (e.g., Bernard-Soulier syndrome, Glanzmann thrombasthenia) or acquired (e.g., due to drugs, alcohol, uremia).
Thrombocytopenia
Thrombocytopenia is defined as a platelet count below 150 x 109/L. Severity is classified as mild (50–150), moderate (20–50), or severe (<20). Bleeding risk increases as platelet count decreases, but clinical bleeding also depends on platelet function and endothelial integrity.
Diagnosis: Full blood count, blood film, and exclusion of congenital causes or secondary factors (e.g., drugs, infections).
Etiology: Includes immune thrombocytopenic purpura (ITP), thrombotic microangiopathies (TTP, HUS, HELLP, HIT), and secondary causes (e.g., drugs, viral infections).
Platelet Function Defects
Bernard-Soulier syndrome: Defect in GP Ib-IX-V, leading to poor platelet adhesion and aggregation.
Glanzmann thrombasthenia: Defect in GP IIb/IIIa, resulting in failure to bind fibrinogen and impaired aggregation.
Primary Haemostasis
Primary haemostasis results in the formation of a platelet plug and does not involve the coagulation pathways. Disorders may affect platelet number, size, or function.
Secondary Haemostasis
Coagulation Cascade
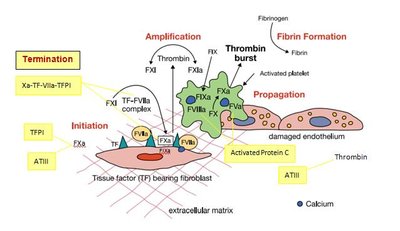
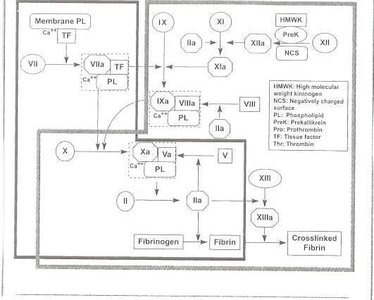
The coagulation cascade consists of the extrinsic, intrinsic, and common pathways, culminating in the formation of a fibrin clot. Fibrinolysis follows, dissolving the clot and preventing excessive thrombosis.
Extrinsic pathway: Initiated by tissue factor (TF) exposure, activating factor VII and subsequently factor X.
Intrinsic pathway: Amplifies the cascade via factors IXa, VIIIa, and X, generating large amounts of thrombin.
Common pathway: Thrombin cleaves fibrinogen to fibrin, which is stabilized by factor XIII.
Fibrinolysis
Fibrinolysis is the process of clot dissolution, primarily mediated by plasmin. Tissue plasminogen activator (t-PA) and urokinase (u-PA) activate plasminogen to plasmin, which degrades fibrin and fibrinogen, releasing fibrin degradation products (FDPs).
Hyperfibrinolysis: Leads to bleeding and thrombosis; regulated by plasmin inhibitors.
Hypofibrinolysis: Associated with thromboembolic disease due to deficiencies in t-PA, u-PA, or plasminogen.
Inherited and Acquired Coagulation Disorders
Inherited Factor Deficiencies
Factor | Defect | Bleeding | Treatment |
|---|---|---|---|
Factor I | Afibrinogenaemia, Hypofibrinogenaemia, Dysfibrinogenaemia | Spontaneous bleeding, abortions, thrombosis | Fibrinogen concentrate, FFP, Cryoprecipitate |
Factor VIII | Haemophilia A (X-linked) | Spontaneous joint/muscle bleeds | Factor VIII concentrates, recombinant prophylaxis |
Factor IX | Haemophilia B | Similar to Haemophilia A | Factor IX concentrates, gene therapy |
Factor XI | Haemophilia C | Bleeding after trauma/surgery | FFP, FXI concentrate |
Factor XIII | Deficiency | Umbilical cord bleeding, delayed wound healing | FXIII concentrate |
Acquired Deficiencies
Liver disease: Affects synthesis of coagulation factors, prolonging PT and APTT.
Vitamin K deficiency: Impairs production of vitamin K-dependent factors (II, VII, IX, X).
Autoantibodies: May develop against factors (e.g., factor VIII inhibitors).
Laboratory Investigation of Haemostasis
Pre-analytical Variables
Venous blood collection, patient identification, anticoagulant use, sample timing, centrifugation, and storage are critical for accurate results.
Laboratory Equipment
Centrifuges, water baths, glass/plastic tubes, pipettes, stopwatches, and automated coagulation analyzers are used for testing.



Manual Tilt Tube Method
This traditional method involves mixing reagent and patient plasma in a glass tube, incubating at 37°C, and timing clot formation. It is now rarely used but demonstrates clot formation to trainees.
Mechanical Coagulometer
Devices like the KC1 micro-mechanical coagulometer detect clot formation by monitoring the movement of a ball bearing in a rotating cuvette. This method is useful for opaque plasma samples.

Screening Tests
Prothrombin Time (PT): Measures the extrinsic pathway (factors VII, V, X, prothrombin, fibrinogen). Normal range depends on instrument and reagent.
Activated Partial Thromboplastin Time (APTT): Measures the intrinsic pathway (factors VIII, IX, XI, XII, I, II, V, X). Sensitive to heparin and lupus anticoagulant.
Fibrinogen (Clauss method): Measures fibrinogen concentration by adding excess thrombin to plasma and timing clot formation.
Further Testing
Thrombin Time (TT), Reptilase Time (RT), Protamine Time: Assess fibrinogen function and presence of inhibitors.
Correction Tests: Mixing patient plasma with normal or adsorbed plasma to distinguish factor deficiencies from inhibitors.
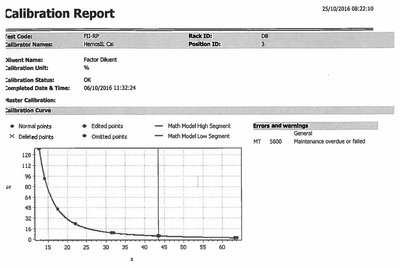
Factor Assays: Quantify specific factor activity using calibration curves.
Dilute Russell Viper Venom Test (DRVVT): Screens for lupus anticoagulant.
Anticoagulant Therapy
Warfarin
Warfarin inhibits vitamin K-dependent carboxylation of coagulation factors, reducing their activity. It is monitored using the PT and International Normalized Ratio (INR):
INR formula:
where PR is the prothrombin ratio (patient/normal clotting time) and ISI is the International Sensitivity Index.

Heparin
Heparin enhances antithrombin III activity, inhibiting thrombin and factor Xa. Low molecular weight heparins are monitored using anti-Xa assays.
Direct Oral Anticoagulants (DOACs)
Thrombin inhibitors: Dabigatran (prolongs PT, APTT, TT).
Factor Xa inhibitors: Rivaroxaban, Apixaban (prolong PT, APTT; TT not affected).
Summary
Haemostasis involves primary (platelet plug) and secondary (coagulation cascade) mechanisms, with regulation by fibrinolysis.
Disorders may be inherited or acquired, affecting platelets or coagulation factors.
Laboratory testing is essential for diagnosis and monitoring of haemostatic disorders and anticoagulant therapy.
