 Back
BackApplied Organometallic Chemistry: Hydrogenation and Hydroelementation Mechanisms
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Applied Organometallic Chemistry
Introduction to Organometallic Catalysis
Organometallic chemistry explores the reactivity of compounds containing metal-carbon bonds, with a focus on their application in catalysis. Transition metal (TM) complexes play a crucial role in activating otherwise inert bonds, enabling transformations such as hydrogenation and hydroelementation. Understanding the mechanisms and selectivity of these reactions is essential for catalyst design and optimization.
Activation Chemistry
Bond Activation by Transition Metals
Activation refers to the process where a transition metal mediates the breakage of a bond that is generally considered inert. This is often achieved through coordination, which weakens the bond and facilitates its cleavage. Hydrogen, while not highly inert, can be activated by TM complexes via different mechanisms:
Homolytic Activation: The H–H bond is split evenly, generating two metal hydride species.
Heterolytic Activation: The H–H bond is cleaved unevenly, producing a hydride and a proton, often stabilized by the metal and a base.
σ-Complexes: These are intermediates where H2 is bound to the metal without full cleavage, as in the Kubas complex.
Example: The Kubas complex, [W(H2)(CO)2(PiPr3)2], was the first crystallized dihydrogen σ-complex, demonstrating the ability of metals to bind and activate H2.
Key Catalytic Cycles: Alkene Hydrogenation
Mechanisms of Alkene Hydrogenation
Hydrogenation of alkenes is a fundamental transformation in organic synthesis, typically catalyzed by transition metal complexes. Two main mechanistic cycles are observed:
Bis-hydride Mechanism: Involves homolytic activation of H2 to form a metal dihydride, followed by alkene coordination, insertion, and reductive elimination to yield the alkane.
Mono-hydride Mechanism: Involves heterolytic activation, often with Ru or Rh complexes, where only one hydride is transferred at a time.
The rate-determining step (RDS) is often the 1,2-insertion of the alkene into the metal-hydride bond, which is a syn-addition process. Steric effects play a significant role in determining the efficiency and selectivity of this step.

Hydrogenation Catalyst Development
Evolution of Catalysts and Turnover Frequency
Hydrogenation catalysts have evolved to improve turnover frequency (TOF), selectivity, and functional group tolerance. Key examples include:
Wilkinson's Catalyst: [RhCl(PPh3)3], a classic homogeneous hydrogenation catalyst.
Schrock-Osborn and Crabtree Catalysts: Offer higher TOFs and broader substrate scope.
Catalyst | TOF (h–1) |
|---|---|
Wilkinson | 13–700 |
Schrock-Osborn | 10–4000 |
Crabtree | 4000–6400 |
Asymmetric Hydrogenation
Chiral Catalysts and Enantioselectivity
Asymmetric hydrogenation enables the synthesis of enantiomerically enriched products using chiral ligands on transition metals. This field was recognized with the 2001 Nobel Prize in Chemistry. The design of chiral phosphine ligands allows for high enantioselectivity in the hydrogenation of prochiral alkenes.
Alkyne Hydrogenation
Mechanism and Selectivity
Alkyne hydrogenation proceeds via similar mechanisms to alkene hydrogenation but requires careful control to avoid over-reduction to alkanes. Selectivity can be tuned by catalyst choice and reaction conditions, allowing for the selective formation of cis- or trans-alkenes.
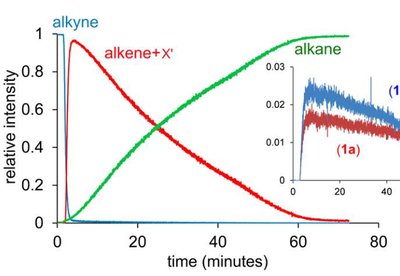
Alternative Hydrogen Activation: Frustrated Lewis Pairs (FLPs)
Metal-Free Hydrogen Activation
Frustrated Lewis pairs (FLPs) are combinations of bulky Lewis acids and bases that cannot form a stable adduct due to steric hindrance. These pairs can heterolytically activate H2 without the need for a transition metal, offering a metal-free route to hydrogenation and related transformations.
Hydroelementation Reactions
Types and Mechanisms
Hydroelementation reactions involve the addition of E–H bonds (E = Si, B, CN, NR2) across unsaturated substrates. Common examples include:
Hydrosilylation: Addition of Si–H to alkenes or alkynes.
Hydroboration: Addition of B–H, typically anti-Markovnikov.
Hydrocyanation: Addition of H–CN.
Hydroamination: Addition of H–NR2.
These reactions often proceed via mechanisms analogous to hydrogenation, with similar catalytic cycles and selectivity considerations.
Hydrosilylation Reactions
Catalytic Cycle and Mechanistic Steps
Hydrosilylation is a key transformation in organosilicon chemistry, catalyzed by transition metals such as Rh or Pt. The catalytic cycle typically involves:
Oxidative addition of Si–H to the metal center
Coordination of the alkene
1,2-Insertion of the alkene into the metal-hydride bond
Reductive elimination to release the product and regenerate the catalyst

Selectivity in Catalysis
Rate- and Selectivity-Determining Steps
The selectivity of catalytic reactions is often determined by the relative activation energies (ΔΔG‡) of competing pathways. The selectivity-determining step may differ from the rate-determining step. Even small differences in activation energy (ca. 3 kcal mol–1) can result in high selectivity for one product over another.
Product Ratio Equation:
Where ΔΔG‡ = ΔG2‡ – ΔG1‡.
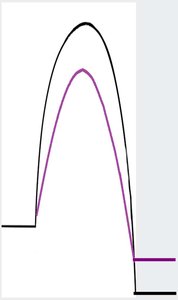
Curtin-Hammett Principle
When two rapidly interconverting intermediates each lead irreversibly to different products, the product ratio depends only on the difference in activation energies (ΔΔG‡), not on their equilibrium concentrations. This principle is crucial for understanding selectivity in hydroelementation and hydrogenation reactions.
Influencing Regioselectivity
Intramolecular hydroelementation (tethering functional groups)
Catalyst design (H-bonding, π-π stacking, steric effects)
Only a small energy difference (~3 kcal mol–1) is needed for full selectivity
Conclusions
Transition metal complexes enable the activation of inert bonds, facilitating key transformations such as hydrogenation and hydroelementation.
Mechanistic understanding and catalyst design are essential for optimizing selectivity and efficiency.
Small differences in activation energy can have a large impact on product selectivity, governed by kinetic control and the Curtin-Hammett principle.
