 Back
BackChimie des Halogénoalcanes, Alcools, Éthers, Thiols et Amines : Substitutions et Éliminations
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Chimie des Halogénoalcanes
Structure et Importance des Halogénoalcanes
Les halogénoalcanes (ou halogénures d’alkyle) sont des composés organiques dans lesquels un ou plusieurs atomes d’hydrogène d’un alcane sont remplacés par des halogènes (F, Cl, Br, I). Bien que rares dans la nature, ils sont essentiels comme intermédiaires de synthèse, solvants, propulseurs, insecticides, anesthésiques, et agents de transport d’oxygène.
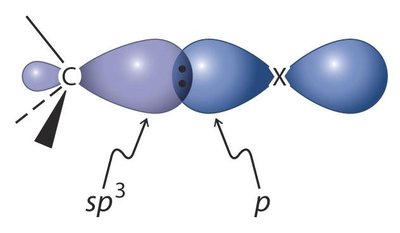
Structure : La liaison C–X résulte du recouvrement d’une orbitale sp3 du carbone et d’une orbitale p de l’halogène.
Polarité : La liaison C–X est polarisée, le carbone étant électrophile (δ+) et l’halogène nucléophile (δ−).
Propriétés Physiques
Les halogénoalcanes présentent des propriétés physiques distinctes des alcanes correspondants, notamment :
Températures d’ébullition : Plus élevées que les alcanes, en raison des interactions dipôle-dipôle et de la polarité accrue.
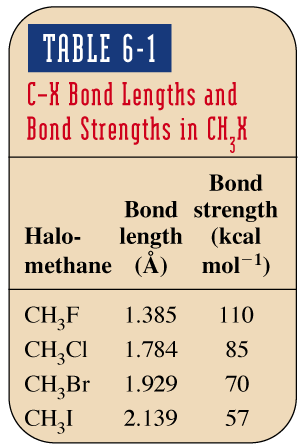
Longueur et force de la liaison C–X : La longueur de la liaison augmente et sa force diminue de F à I.
Halométhane | Longueur de liaison (Å) | Force de liaison (kcal/mol) |
|---|---|---|
CH3F | 1.385 | 110 |
CH3Cl | 1.784 | 85 |
CH3Br | 1.929 | 70 |
CH3I | 2.139 | 57 |
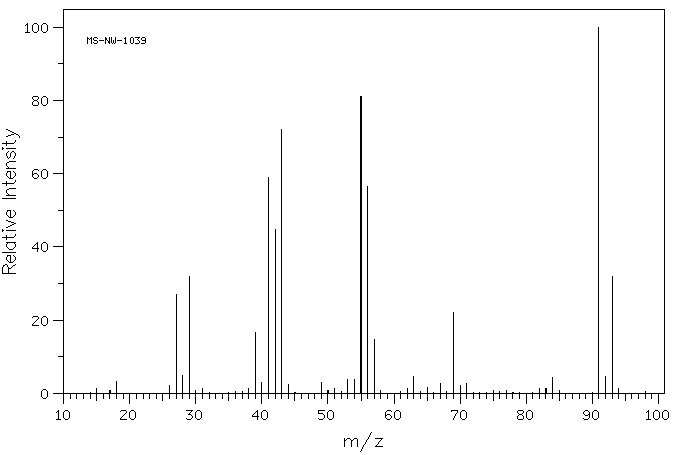
Spectroscopie des Halogénoalcanes
Les halogénoalcanes présentent des signatures caractéristiques en RMN, IR et spectrométrie de masse :
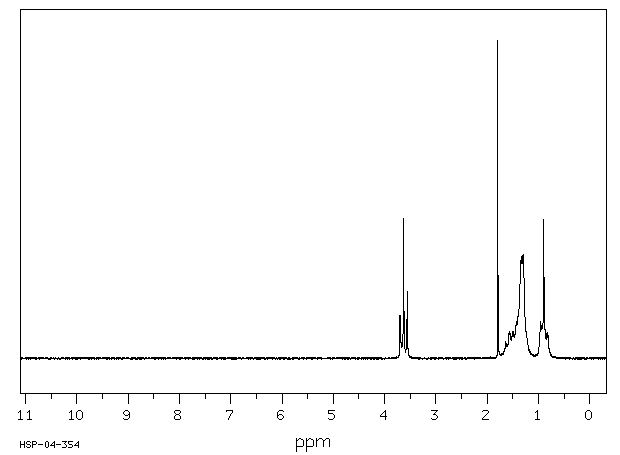
1H-RMN : Les halogènes provoquent un déblindage des protons voisins (3–4 ppm pour Cl).
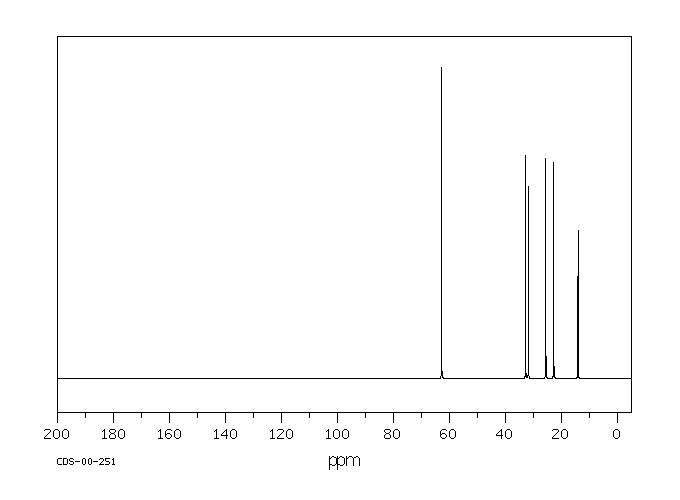
13C-RMN : Les carbones liés à un halogène apparaissent entre 40 et 50 ppm (pour Cl).
IR : Les vibrations C–X apparaissent à des longueurs d’onde plus basses pour les halogènes lourds.
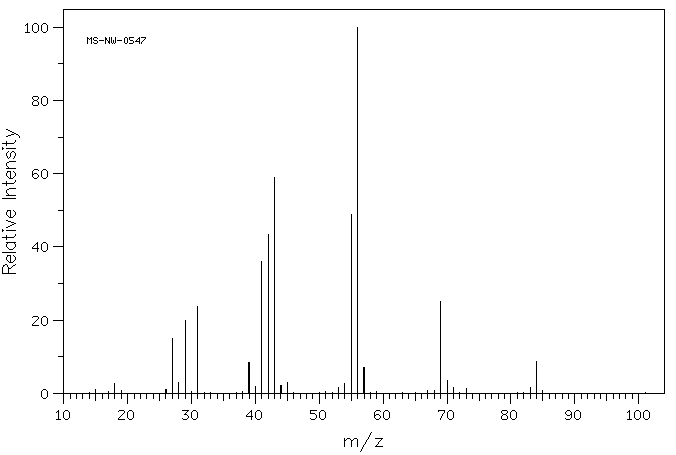
MS : Les isotopes du chlore et du brome permettent d’identifier les fragments spécifiques.
Réactions de Substitution Nucléophile
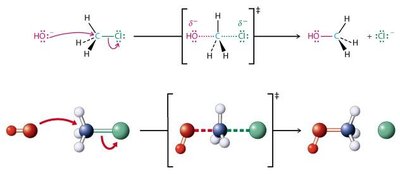
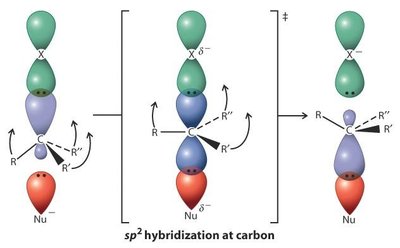
Mécanismes SN2 et SN1
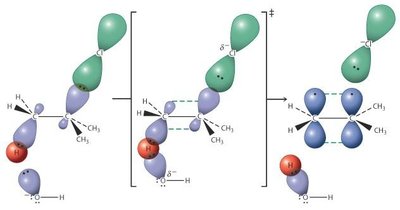
Les halogénoalcanes subissent deux grands types de substitutions nucléophiles : SN2 (bimoléculaire) et SN1 (unimoléculaire).
SN2 : Réaction concertée, inversion de configuration, cinétique d’ordre 2 ().
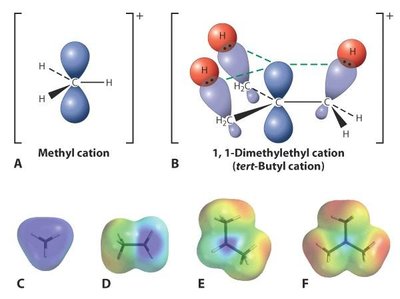
SN1 : Formation d’un carbocation intermédiaire, racémisation possible, cinétique d’ordre 1 ().
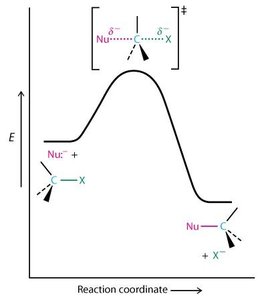
Facteurs Affectant la Réactivité SN2
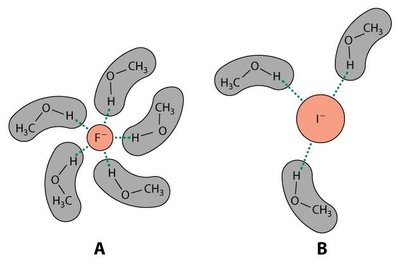
Nucléophile : Plus la charge négative, la basicité et la polarisabilité sont élevées, plus la nucléophilie est forte.
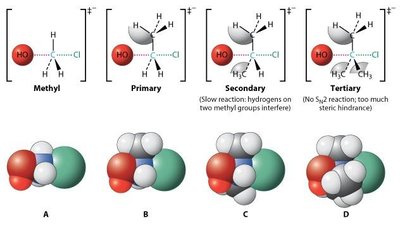
Substrat : Les halogénoalcanes primaires réagissent plus vite que les secondaires, les tertiaires sont inertes (encombrement stérique).
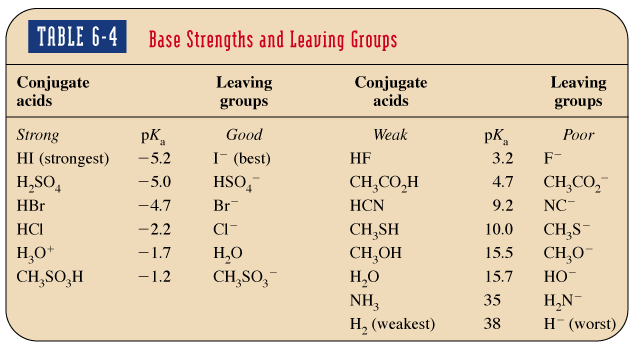
Groupe partant : Un bon groupe partant est la base conjuguée d’un acide fort (I− > Br− > Cl− > F−).
Solvant : Les solvants polaires aprotiques favorisent la SN2.
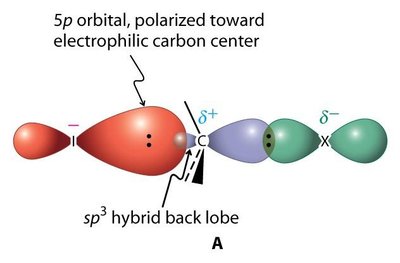
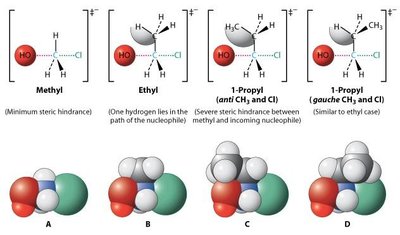
Facteurs Affectant la Réactivité SN1
Substrat : Les halogénoalcanes tertiaires réagissent le plus vite (stabilisation du carbocation par hyperconjugaison).
Solvant : Les solvants polaires protiques accélèrent la SN1.
Nucléofuge : Un excellent groupe partant favorise la SN1.
Nucléophile : N’influence pas la vitesse de la SN1.
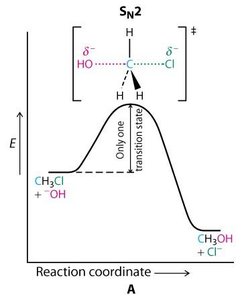
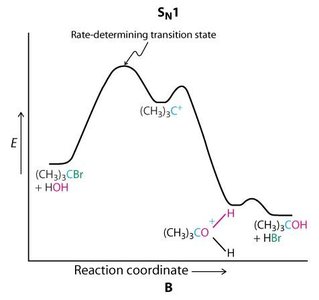
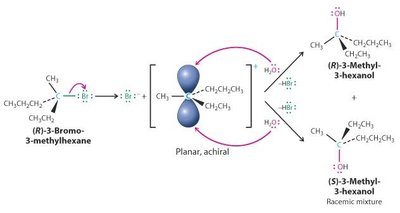
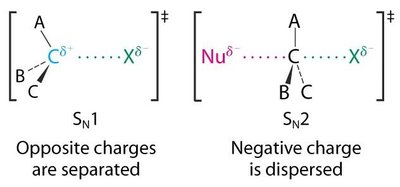
Réactions d’Élimination : E1 et E2
Mécanismes E1 et E2
Les halogénoalcanes peuvent aussi subir des réactions d’élimination pour former des alcènes :
E1 : Formation d’un carbocation intermédiaire, puis départ d’un proton. Favorisée par substrats tertiaires, solvants polaires protiques, bases faibles.
E2 : Déprotonation et départ du groupe partant en une seule étape, stéréospécifique (anti-coplanarité), favorisée par bases fortes et substrats encombrés.
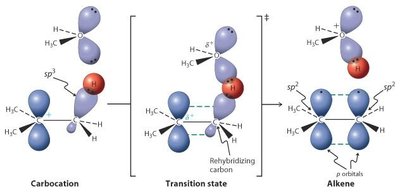
Régio- et Stéréosélectivité des Éliminations
Règle de Saytzeff : L’alcène le plus substitué est généralement favorisé.
Règle de Hofmann : Avec des bases encombrées, l’alcène le moins substitué est favorisé.
Stéréospécificité : En E2, la géométrie anti est requise pour l’élimination.
Compétition Substitution/Élimination
Le mécanisme préféré dépend de la nature du substrat, du nucléophile/base, et du solvant :
Type d’halogénoalcane | Nucléophile faible | Nucléophile fort | Base forte |
|---|---|---|---|
Méthyle | Pas de réaction | SN2 | SN2 |
Primaire | Pas de réaction | SN2 | E2 |
Secondaire | SN1/E1 | SN2/E2 | E2 |
Tertiaire | SN1/E1 | E2 | E2 |

Chimie des Alcools
Structure, Propriétés et Nomenclature
Les alcools sont des composés organiques contenant un groupe hydroxyle (–OH) lié à un carbone sp3. Ils sont nommés par le suffixe –ol.
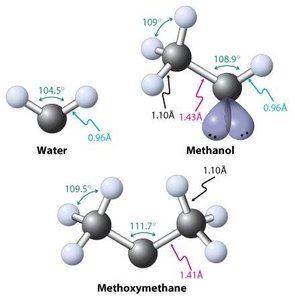
Structure : L’oxygène est sp3 hybridé, l’angle de liaison est proche de 109°.
Liaison O–H : Plus courte et plus polarisée que C–H, favorisant les liaisons hydrogène.
Solubilité : Les alcools sont solubles dans l’eau grâce aux liaisons hydrogène.
Acidité : pKa ≈ 15–18, plus acides que les alcanes mais moins que les acides carboxyliques.
Spectroscopie des Alcools
1H-RMN : Le proton de l’alcool est variable (3–4 ppm), large à cause de l’échange de protons.
13C-RMN : Les carbones porteurs d’un OH apparaissent entre 50 et 80 ppm.
IR : Bande O–H caractéristique entre 3200–3500 cm−1.
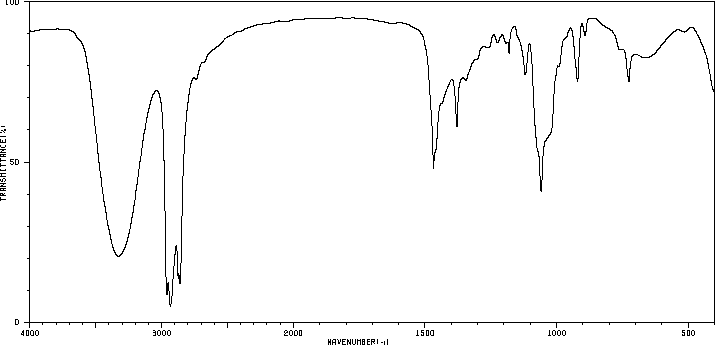
Préparation des Alcools
Fermentation : Production d’éthanol à partir de sucres.
Réduction des composés carbonylés : Utilisation de NaBH4 ou LiAlH4 pour réduire aldéhydes et cétones en alcools.
Addition de réactifs de Grignard : Formation d’alcools par addition d’organomagnésiens sur des composés carbonylés.

Chimie des Éthers
Structure, Nomenclature et Propriétés
Les éthers sont des composés de formule générale R–O–R'. Ils sont nommés comme des alcanes portant un substituant alkoxy ou en nommant les deux groupes suivis de «éther».
Points d’ébullition : Plus bas que les alcools correspondants (pas de liaisons hydrogène entre molécules d’éther).
Polyéthers cycliques : Les éthers couronnes peuvent solubiliser des cations métalliques par coordination.
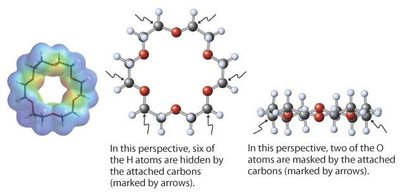
Synthèse des Éthers
Williamson : Réaction SN2 entre un alcoolate et un halogénoalcane primaire.
Déshydratation d’alcools : Formation d’éthers symétriques en présence d’acide fort.
Chimie des Thiols et Sulfures
Structure, Propriétés et Réactivité
Les thiols (R–SH) et sulfures (R–S–R') sont les analogues soufrés des alcools et éthers. Le soufre, plus gros et plus polarisable que l’oxygène, confère aux thiols une acidité supérieure (pKa ≈ 10–12) et une meilleure nucléophilie.


Chimie des Amines
Structure, Propriétés et Synthèse
Les amines (R–NH2, R2NH, R3N) sont des composés azotés basiques et nucléophiles. Elles sont nommées par le suffixe –amine ou par des noms triviaux. Les amines primaires peuvent être préparées par réduction de nitriles, azotures ou par la synthèse de Gabriel.

Résumé des Concepts à Maîtriser
Nomenclature des halogénoalcanes, alcools, thiols et amines
Mécanismes SN2, SN1, E1, E2 et facteurs influençant leur occurrence
Compétition entre substitution et élimination
Réduction des carbonyles, réactions de Grignard
Activation douce des alcools, oxydation, chimie des éthers, ouverture des époxydes
Nucléophilicité et oxydation des composés soufrés
Synthèse des amines primaires
