 Back
BackGenomics, Bioinformatics, and Proteomics: Foundations and Applications
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Genomics, Bioinformatics, and Proteomics
Introduction to Genomics and Bioinformatics
Genomics is the comprehensive study of genomes, encompassing the structure, function, evolution, and mapping of genetic material in organisms. Bioinformatics applies mathematical and computational tools to organize, analyze, and interpret vast amounts of genetic data, including gene structure, sequence, expression, and protein function.
Genomics provides insights into the complete DNA content of organisms, enabling comparative and functional studies.
Bioinformatics is essential for managing and analyzing the large datasets generated by genomic research.
Whole-Genome Sequencing (WGS)
Whole-genome sequencing (WGS), also known as shotgun sequencing, is the primary strategy for sequencing and assembling entire genomes. This method involves fragmenting genomic DNA, sequencing the fragments, and using computational tools to assemble the genome.
Step 1: Genomic DNA is cut with restriction enzymes to create overlapping fragments.
Step 2: Overlapping fragments are sequenced.
Step 3: Computer programs align fragments based on identical DNA sequences, forming contiguous sequences (contigs).
Step 4: Contigs are assembled to reconstruct the entire chromosome.
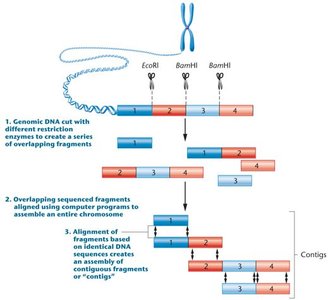
Example: Digestion with EcoRI and BamHI produces overlapping fragments that are aligned to assemble the chromosome.
Bioinformatics Applications
Bioinformatics uses algorithm-based software to align DNA sequences, identify overlapping regions, and reconstruct chromosome order. It is also used to:
Compare DNA sequences
Identify genes and regulatory regions (promoters, enhancers)
Predict amino acid sequences
Delineate evolutionary relationships between genes
Contigs and Sequence Alignment
Contigs are continuous DNA fragments formed by aligning overlapping sequences. Sequence alignment is crucial for assembling genomes and identifying gene locations.
Alignment involves lining up similar sequences for comparison.
Contigs collectively form a continuous DNA molecule within a chromosome.
GenBank and Sequence Databases
GenBank, maintained by the National Center for Biotechnology Information (NCBI), is the largest public DNA sequence database. Each sequence receives a unique accession number for retrieval and analysis.
BLAST: Basic Local Alignment Search Tool
BLAST is a software application used to compare newly sequenced DNA to known sequences in databases. It calculates:
Identity value: Proportion of identical matches between aligned sequences.
E-value: Statistical measure of the likelihood that the match occurred by chance.

Example: A rat chromosome 12 contig aligned with a mouse chromosome 8 sequence shows 93% identity, indicating evolutionary conservation.
Gene Annotation and Hallmark Features
Gene annotation identifies hallmark features of genes, such as exons, introns, regulatory sequences (promoters, enhancers, silencers), untranslated regions (UTRs), and termination sequences. Distinguishing these elements is essential for understanding gene structure and function.

Open Reading Frames (ORFs)
Open reading frames (ORFs) are sequences of nucleotides that can be translated into proteins. ORFs typically begin with an initiation codon (ATG) and end with a stop codon (TAA, TAG, TGA).
In eukaryotes, ORFs include both exons and introns.
Identifying ORFs is a key step in gene prediction.
Predicting Gene and Protein Functions
Functional genomics interprets DNA sequences to establish gene functions, often using experimental approaches to confirm computational predictions. Sequence similarity searches (e.g., BLAST) can infer gene function based on homology to known genes.
Genes with similar sequences are likely to encode proteins with similar functions.
Homologous Genes: Orthologs and Paralogs
Homologous genes are evolutionarily related. Orthologs are genes in different species that descended from a common ancestor. Sequence comparisons reveal evolutionary conservation and functional similarities.
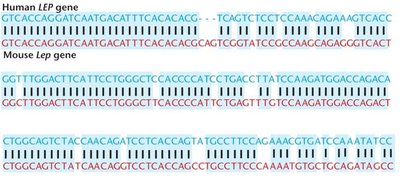
Example: The human LEP gene and mouse Lep gene show high sequence similarity, indicating conserved function.
Protein Domains and Motifs
Gene sequences can predict polypeptide sequences, which are analyzed for specific protein domains and motifs. These structural features help predict protein function (e.g., ion channels, membrane-spanning regions, DNA-binding motifs like helix-turn-helix, leucine zipper, zinc-finger).
Major Features of the Human Genome
Feature | Description |
|---|---|
Genome Size | ~3.1 billion nucleotides |
Protein-coding DNA | ~2% of genome |
Gene Number | ~20,000 protein-coding genes |
Gene Diversity | Alternative splicing produces up to 200,000 proteins |
Genome Similarity | 99.9% identical among humans |
Genomic Variation | SNPs and CNVs account for diversity |
Repetitive DNA | ~50% from transposable elements |
Gene Distribution | Non-uniform; gene-rich and gene-poor regions |
Additional info: Chromosome 19 has the highest gene density; chromosome 13 and Y have the lowest.
Major Features of the Human Genome Project (HGP)
The HGP revealed that less than 2% of the genome codes for proteins and that there are about 20,000 protein-coding genes. Alternative splicing allows for a much greater diversity of proteins than the number of genes alone would suggest.
Functional Categories of Genes
Genes are categorized based on known or predicted functions, sequence similarity to genes in other species, and analysis of protein domains and motifs.
Individual Variations in the Human Genome
Most human genetic variation arises from:
Single-nucleotide polymorphisms (SNPs): Single-base changes associated with disease and traits.
Copy number variations (CNVs): DNA segments that are duplicated or deleted.
Accessing the Human Genome Project
Genome databases provide maps for all human chromosomes, aiding in the identification of disease genes and the development of new treatments.

Omics Disciplines
"Omics" refers to large-scale studies of biological molecules, including:
Proteomics
Metabolomics
Glycomics
Toxicogenomics
Metagenomics
Pharmacogenomics
Transcriptomics
Whole-Exome Sequencing (WES)
WES sequences only the exons (protein-coding regions) of the genome, identifying mutations relevant to disease. However, it does not detect regulatory region mutations.
ENCODE Project
The Encyclopedia of DNA Elements (ENCODE) uses experimental and computational approaches to identify functional elements in the genome, such as transcription start sites, promoters, and enhancers.
Nutrigenomics
Nutrigenomics studies the interaction between nutrition and genes, providing personalized dietary recommendations based on genetic makeup.
Stone-Age Genomics
Stone-age genomics analyzes ancient DNA from fossils to study evolutionary relationships among extinct and modern species.
Comparative Genomics
Comparative genomics compares genomes across species to discover genes, study evolution, and understand organism-environment interactions.
Organism | Genome Size | Chromosome Number | Gene Number | % Genes Shared with Humans |
|---|---|---|---|---|
Human | 3.1 Gb | 46 | ~20,000 | 100 |
Chimpanzee | 3 Gb | 48 | ~20,000-24,000 | 98 |
Mouse | ~2.5 Gb | 40 | ~30,000 | 80 |
Yeast | 12 Mb | 32 | ~5,700 | 30 |
Fruit fly | 165 Mb | 8 | ~13,600 | 50 |
Rice | 389 Mb | 24 | ~41,000 | Not determined |
The Neanderthal Genome and Modern Humans
Sequencing of Neanderthal DNA revealed that modern non-African humans have 1–4% Neanderthal DNA, indicating interbreeding between the species 45,000–80,000 years ago. Comparative genomics identified regions of rapid human evolution since divergence from Neanderthals.
Metagenomics
Metagenomics uses WGS to sequence genomes from environmental samples, revealing the diversity of microbial communities without the need for culturing. Studies have shown that many environmental DNA sequences do not match known organisms.
The Human Microbiome Project (HMP)
The HMP aims to sequence the genomes of microorganisms living in and on humans. Metagenomic analysis of the human microbiome has identified unique and shared microbial genes associated with various diseases.
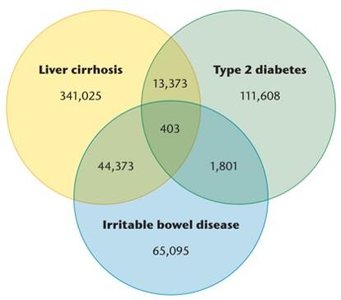
Example: A Venn diagram shows overlapping and unique gut microbial genes in liver cirrhosis, type 2 diabetes, and irritable bowel disease.
Proteomics
Proteomics is the large-scale study of the complete set of proteins (proteome) encoded by a genome. It provides information on protein structure, function, interactions, localization, and modifications, and is crucial for identifying disease biomarkers.
Proteomics Technologies: Two-Dimensional Gel Electrophoresis (2DGE)
2DGE separates proteins based on isoelectric point (pH) and molecular weight, allowing high-resolution analysis of complex protein mixtures.
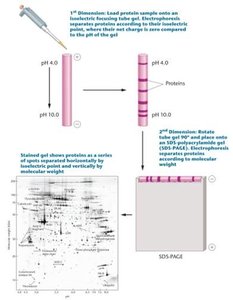
Example: Human platelet proteins separated by 2DGE, with each spot representing a different polypeptide.
Proteomics Technology: Mass Spectrometry
Mass spectrometry (MS) analyzes ionized protein samples to determine their mass-to-charge ratio, enabling identification and characterization of proteins. Matrix-assisted laser desorption ionization (MALDI) is a common MS technique in proteomics.

Example: MS identifies the amino acid sequence of a peptide isolated from a 2D gel.
