 Back
BackGenomics: Genetics from a Whole-Genome Perspective (Chapter 16) – Study Notes
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Genomics: Genetics from a Whole-Genome Perspective
Introduction to Genomics
Genomics is the study of the structure, function, evolution, and mapping of entire genomes. It provides insights into the organization, content, and function of genetic material in various organisms, with a focus on large-scale, comparative, and functional analyses.
Genome Size and Gene Content
Variation in Genome Size
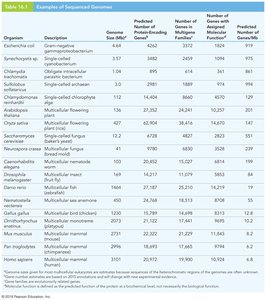
Eukaryote genomes are generally larger and contain more genes than those of eubacteria and archaea.
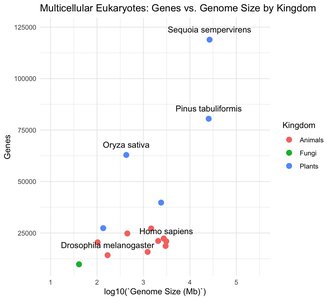
Multicellular organisms tend to have larger genomes and more genes, but there is extreme variation in genome size, especially among plants due to polyploidy.
Gene number for multicellular eukaryotes ranges from ~10,000 to ~100,000, with higher numbers often due to polyploidy.
Most genome size variation is not due to differences in gene number, but rather to repetitive elements and non-coding DNA.
Genes and mRNAs
Genes are composed of exons (coding regions) and introns (non-coding regions removed during RNA splicing).
Exons are retained in mature mRNA, while introns are removed.
Untranslated regions (UTRs) are parts of exons that are not translated into protein.
The coding sequence (CDS) is the portion of exons that is translated into protein.
Genome Composition
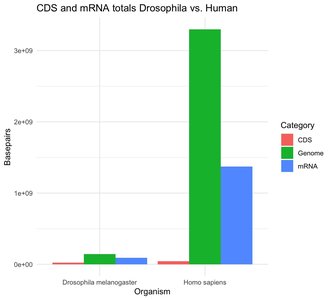
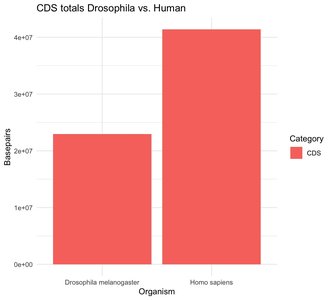
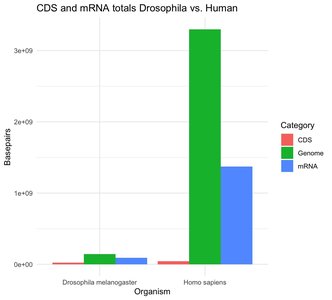
Large amounts of genomic DNA are not transcribed into mRNA (e.g., ~67% in humans, ~40% in Drosophila).
CDS makes up a very small proportion of the genome and mRNA.
Introns can be very large, especially in primates, while plants generally have small introns.
Most genome size variation is due to repetitive elements, not differences in CDS.
Reasons for Genome Size Variation
Repetitive elements, transposons, and transposon fossils (junk DNA) are the major contributors to genome size variation.
Polyploidy and genome duplication also increase genome size.
Introns vary in size between species.
Differences in gene number are a minor reason for genome size variation.
DNA Sequencing Methods
Parameters of Sequencing Methods
Cost per base and per run
Clonal (requires purified DNA) vs. parallel (can sequence mixtures)
Accuracy, read length, and reads per run
Common methods include Sanger sequencing, Illumina SBS, Pacific Biosciences, and Oxford Nanopore, each with different strengths and applications.
Genome Sequencing and Assembly
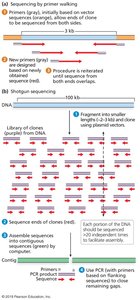
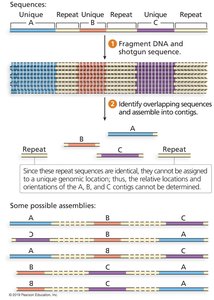
Sequencing reads are much shorter than chromosomes, making assembly challenging, especially in repetitive regions.
Paired-end sequencing and long-read technologies (e.g., PacBio, Oxford Nanopore) have improved assembly quality.
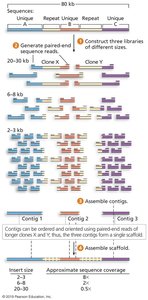
Genome assembly involves organizing reads into contigs and scaffolds.
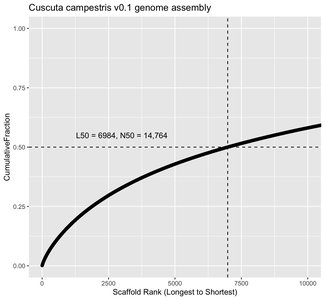
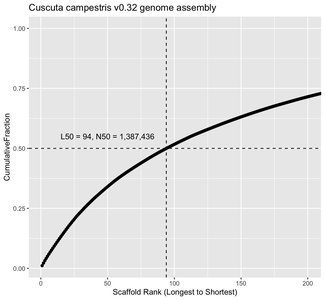
Genome Assembly Metrics
L50: The number of scaffolds such that 50% of the genome is contained in scaffolds of this length or longer.
N50: The length of the scaffold at the L50 rank.
Lower L50 and higher N50 indicate better assembly quality.
Genome Annotation
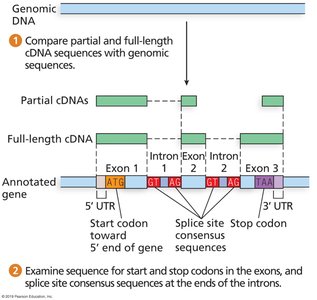
Annotation Process
Annotation identifies genes, exons, introns, UTRs, promoters, enhancers, and repeats.
Experimental methods (e.g., RNA-seq) and computational predictions are used.
Challenges include alternative splicing, alternative start sites, and uncertain CDS boundaries.
Functional Annotation
Assigns function to genes based on experiments or homology.
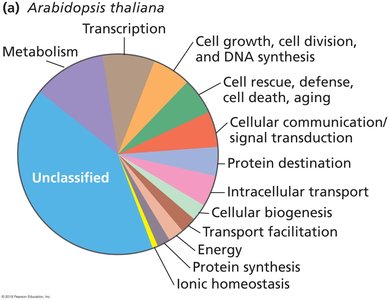
Many proteins remain unclassified or of unknown function.
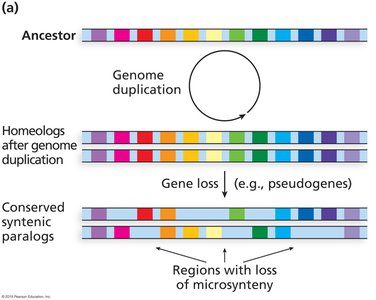
Genome Organization and Synteny
Genome Organization
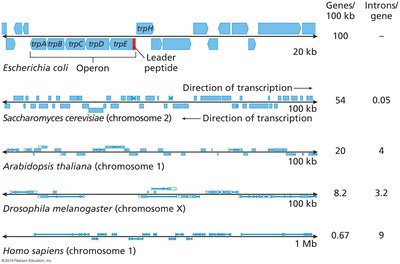
Genes and repetitive elements are interspersed throughout the genome.
Gene density and intron number vary widely among species and chromosomes.
Synteny
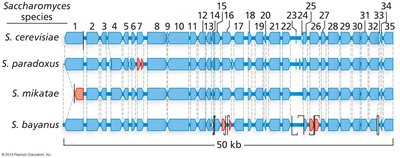
Synteny is the conserved order of genes among related species.
It is more easily detected in animals than in plants due to fewer polyploidy events in animals.
Evolution of Genomes
How New Genes Are Born
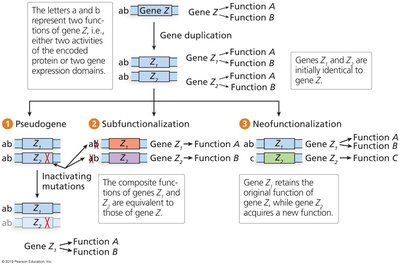
Exon shuffling, retrotransposition, and horizontal gene transfer contribute to new gene formation.
Transposable elements (TEs) can be co-opted into gene function.
Gene duplication can lead to pseudogenes, subfunctionalization, or neofunctionalization.
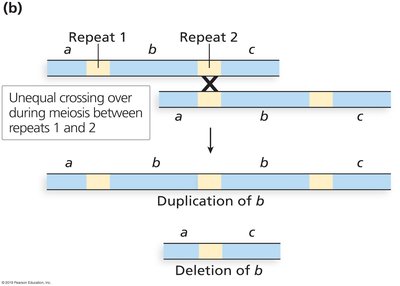
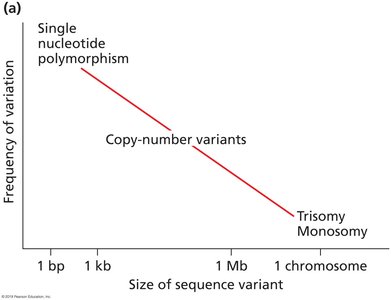
Genome Variation and Copy Number Variation
Heterozygosity, copy number variants, and aneuploidies complicate genome assembly and analysis.
Mispairing of tandem repeats during meiosis can generate copy number variation.
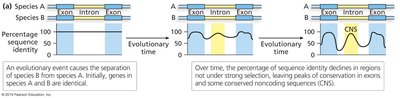
Comparative Genomics and Phylogenetic Footprinting
Phylogenetic Footprinting
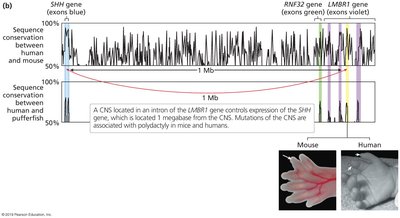
Comparing syntenic regions across species reveals conserved sequences, especially in coding regions (CDS).
Phylogenetic footprinting is effective in animals but less so in plants due to polyploidy and genome fractionation.
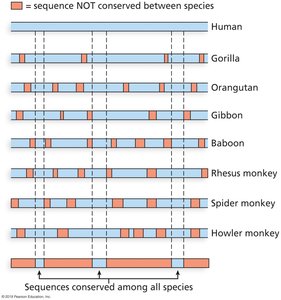

Applications of Comparative Genomics
Identification of regulatory elements (e.g., enhancers) by conserved non-coding sequences.
Understanding evolutionary relationships and genome evolution.
Additional info: These notes integrate and expand upon the provided materials, including definitions, examples, and explanations of key genomics concepts, as well as relevant images and tables to reinforce understanding.
