 Back
BackThalassemia: Genetic Basis, Classification, and Clinical Features
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Thalassemia: Overview
Definition and Origin
Thalassemia refers to a group of inherited blood disorders characterized by reduced or absent synthesis of one or more globin chains of hemoglobin. The term is derived from the Greek word thalassa meaning "sea," reflecting its high prevalence in populations around the Mediterranean Sea.
Classification of Thalassemia
Globin Chain Defects
Thalassemias are classified based on which globin chain is affected:
Alpha (α)-thalassemia: Reduced or absent α-globin chain synthesis
Beta (β)-thalassemia: Reduced or absent β-globin chain synthesis
Gamma (γ)-thalassemia: Reduced or absent γ-globin chain synthesis
Delta (δ)-thalassemia: Reduced or absent δ-globin chain synthesis
Gamma-delta-beta (γδβ)-thalassemia: Reduced or absent γ, δ, and β-globin chains
The most common types are α-thalassemia and β-thalassemia.
Hemoglobin Structure and Genetics
Hemoglobin Composition
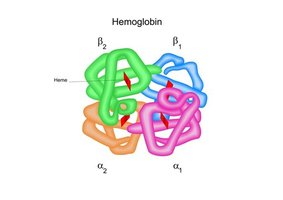
Hemoglobin is a tetramer composed of two α and two β globin chains. The proper synthesis of these chains is essential for normal oxygen transport.
Genetic Location of Globin Genes
The α-globin genes are located on chromosome 16, while the β-globin gene is located on chromosome 11. Each person has four α-globin genes (two per chromosome 16) and two β-globin genes (one per chromosome 11).

Beta (β)-Thalassemia
Genetic Basis and Types
β-thalassemia is caused by mutations in the β-globin gene on chromosome 11, resulting in absent (β⁰) or reduced (β⁺) β-globin production. Types include:
β-Thalassemia minor (trait): Heterozygous, mild anemia
β-Thalassemia intermedia: Moderate anemia, variable clinical course
β-Thalassemia major (Cooley’s anemia): Severe anemia, transfusion-dependent
Pathophysiology
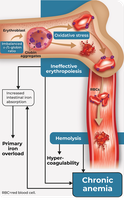
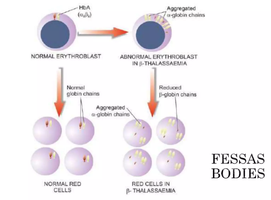
Reduced β-globin chains lead to an excess of α chains, which are unstable and precipitate as Fessas bodies. These damage red cell membranes, causing destruction of red cell precursors in the bone marrow (ineffective erythropoiesis) and mature red cells in the spleen.
β-Thalassemia Minor (Trait)
In β-Thalassemia Minor, one β-globin gene is mutated, leading to reduced synthesis of β chains. The remaining normal gene produces enough β chains for functional hemoglobin, resulting in only mild anemia.
Laboratory Findings
CBC: Mild microcytic, hypochromic anemia
MCV: Slightly reduced (60–75 fL)
Peripheral smear: Target cells, slight anisopoikilocytosis
Hemoglobin electrophoresis:
HbA (α₂β₂): Slightly decreased
HbA₂ (α₂δ₂): Increased (>3.5%, usually 4–7%)
HbF (α₂γ₂): Normal or slightly increased
Genetic testing: Heterozygous β-globin gene mutation
β-Thalassemia Intermedia
Mutations affect both β-globin genes, but at least one produces some β chains. Genotypes include heterozygous (β⁰/β⁺) and homozygous (β⁺/β⁺).
Clinical features: Moderate anemia, splenomegaly, delayed growth, mild skeletal changes
Laboratory findings:
CBC: Moderate microcytic hypochromic anemia, Hb 6–10 g/dL
Peripheral smear: Target cells, poikilocytosis, basophilic stippling, NRBCs
Hemoglobin electrophoresis:
HbA: Decreased (but present)
HbA₂: Moderately to markedly increased (10–50%)
HbF: Mildly increased (>3.5%)
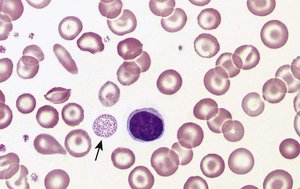
β-Thalassemia Major (Cooley’s Anemia)
Both β-globin genes are mutated (β⁰/β⁰), leading to absent or severely reduced β-globin chains. Hemoglobin A cannot form properly, causing severe anemia and α-chain precipitation.
Symptoms: Severe anemia, failure to thrive, jaundice, bone deformities, hepatosplenomegaly, frequent infections
Laboratory findings:
CBC: Very low hemoglobin (3–6 g/dL), microcytic, hypochromic anemia, increased RDW
Peripheral smear: Target cells, nucleated RBCs, anisopoikilocytosis
Hemoglobin electrophoresis:
HbF: Markedly increased (>90%)
HbA: Absent or very low
HbA₂: Variable
Iron studies: May show iron overload due to frequent transfusions
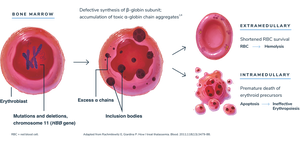
Other Beta-Cluster Gene Defects
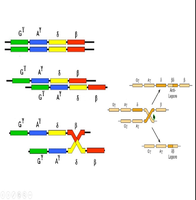
Hemoglobin Lepore
Hemoglobin Lepore is a structural variant resulting from unequal crossing-over between δ- and β-globin genes, producing a δβ fusion chain. It behaves as a β-thalassemia–like disorder due to reduced β-globin production.
Alpha (α)-Thalassemia
Molecular Basis
Alpha thalassemia is caused by reduced or absent production of α-globin chains, leading to an imbalance with β chains in adults and γ chains in fetuses. The severity depends on how many α genes are affected.

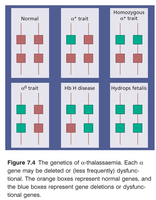
Classification by Gene Deletion
The clinical severity is classified according to the number of missing or inactive α genes:
Genotype | Clinical State | Symptoms |
|---|---|---|
αα/αα | Normal | None |
-α/αα | Silent carrier | None, usually healthy |
-α/-α or --/αα | Minor (trait) | Mild anemia |
--/-α | Hb H disease | Moderate to severe anemia, jaundice, splenomegaly |
--/-- | Bart’s hydrops fetalis | Severe fetal anemia, heart failure, usually lethal |
Silent Carrier State
One α gene missing. No symptoms, normal or very mild anemia. Usually discovered by family studies.
Alpha Thalassemia Trait
Two α genes missing (can be on same or different chromosomes). Mild anemia, often mistaken for iron deficiency.
Hemoglobin H Disease
Three α genes missing. Only one gene produces α chains, resulting in excess β chains forming β₄ tetramers (hemoglobin H). Hemoglobin H is unstable and precipitates within red cells, leading to hemolysis in the spleen.
Symptoms: Moderate to severe anemia, jaundice, enlarged spleen
Laboratory findings: Microcytic, hypochromic RBCs, marked poikilocytosis, numerous target cells, Heinz-like bodies (damaged hemoglobin clumps)
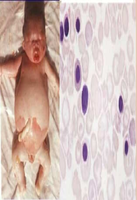
Bart’s Hydrops Fetalis Syndrome
Most severe form, incompatible with life. No functioning α chain genes (--/--). Severe fetal anemia, heart failure, often stillbirth. Formation of Hb Bart’s (γ₄ tetramers) results in very low oxygen delivery.
Summary Table: Alpha Thalassemia Genotypes and Clinical States
Genotype | Clinical State | Symptoms |
|---|---|---|
αα/αα | Normal | None |
-α/αα | Silent carrier | None |
-α/-α or --/αα | Minor (trait) | Mild anemia |
--/-α | Hb H disease | Moderate to severe anemia |
--/-- | Bart’s hydrops fetalis | Severe fetal anemia, lethal |
Key Concepts and Equations
Hemoglobin composition:
Hemoglobin electrophoresis: Used to distinguish thalassemia types by quantifying HbA, HbA₂, and HbF
Genetic inheritance: Thalassemia is inherited in an autosomal recessive manner
Additional info: Thalassemia is a classic example of a genetic disorder affecting gene regulation, protein synthesis, and chromosomal variation, making it highly relevant to genetics courses.
