 Back
BackAlkene Reactivity: Controlled Hydration, Dihydroxylation, and Oxidative Cleavage
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Alkene Reactivity
Introduction
Alkenes are versatile organic compounds that undergo a variety of addition reactions. This section focuses on the regiochemical and stereochemical control in alkene hydration, dihydroxylation, and oxidative cleavage, as well as additional electrophilic addition reactions. Understanding these mechanisms is crucial for predicting product outcomes and designing synthetic routes in organic chemistry.
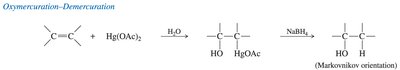
Oxymercuration–Demercuration
Overview and Purpose
Oxymercuration–demercuration is a two-step method for hydrating alkenes to produce alcohols with Markovnikov regioselectivity, while avoiding carbocation rearrangements. This reaction provides better control over product structure compared to direct acid-catalyzed hydration.
Step 1: Oxymercuration – Alkene reacts with mercuric acetate, forming a mercurinium ion intermediate.
Step 2: Demercuration – Sodium borohydride (NaBH4) reduces the organomercury intermediate, yielding the alcohol.
Markovnikov Orientation – The hydroxyl group attaches to the more substituted carbon.
Mechanism of Oxymercuration
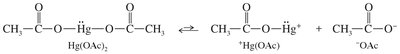
Mercuric Acetate Dissociation: dissociates to form an electrophilic species.
Mercurinium Ion Formation: The alkene attacks , forming a three-membered mercurinium ion.
Nucleophilic Attack: Water (or alcohol) opens the ring, adding to the more substituted carbon (Markovnikov addition).
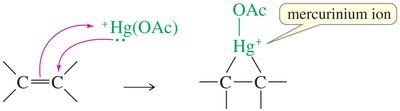
Stereochemistry of Mercurinium Ion Opening
The nucleophile attacks the more substituted carbon from the side opposite to mercury (anti addition).
Demercuration
Sodium borohydride (NaBH4) replaces the mercury group with hydrogen, completing the formation of the alcohol.
The overall process does not involve carbocation rearrangement, preserving the carbon skeleton.
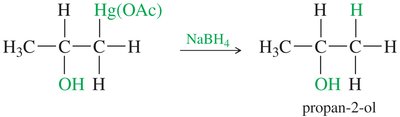
Example: Oxymercuration–Demercuration
Application to substituted alkenes demonstrates Markovnikov selectivity and high yield.
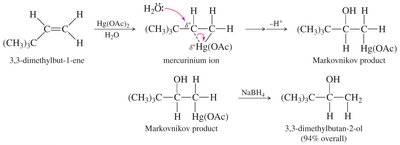
Alkoxymercuration–Demercuration
Ether Formation
When an alcohol is used instead of water as the nucleophile, the reaction yields an ether instead of an alcohol. This is useful for synthesizing asymmetric ethers.
Hydroboration–Oxidation
Overview and Purpose
Hydroboration–oxidation is a two-step reaction that hydrates alkenes with anti-Markovnikov regioselectivity, placing the hydroxyl group on the less substituted carbon. This method also avoids carbocation rearrangements and proceeds with syn addition.
Step 1: Hydroboration – Borane (BH3) adds across the double bond.
Step 2: Oxidation – Hydrogen peroxide (H2O2) and base convert the organoborane to an alcohol.
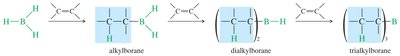
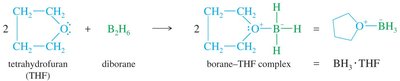
Diborane and Borane–THF Complex
Diborane () is in equilibrium with borane ().
Borane is stabilized in solution by complexation with tetrahydrofuran (THF).
Mechanism of Hydroboration
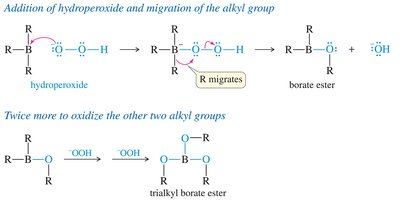
Borane adds to the less hindered carbon of the alkene, resulting in syn addition of hydrogen and boron.
Multiple additions can occur, forming trialkylboranes.
Oxidation to Alcohol
Hydroperoxide ion () is the active oxidant.
Oxidation replaces the boron atom with a hydroxyl group, retaining the syn stereochemistry.
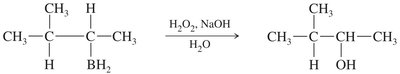
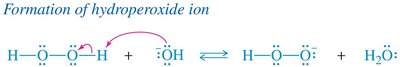
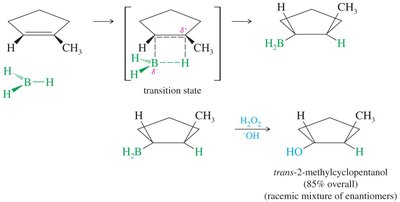
Stereochemistry of Hydroboration–Oxidation
Both hydrogen and hydroxyl are added to the same face of the alkene (syn addition).
Product is often a racemic mixture if a new stereocenter is formed.
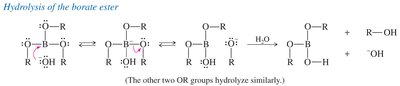
Hydrolysis of Borate Ester
Hydrolysis with base releases the alcohol from the borate ester.
Epoxidation and Dihydroxylation of Alkenes
Epoxidation
Epoxidation converts an alkene into an epoxide (oxirane) using a peroxyacid (e.g., mCPBA). Epoxides are reactive intermediates that can be further transformed into diols.
Epoxidation is a concerted, stereospecific reaction.

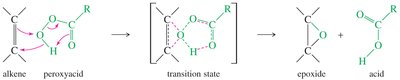
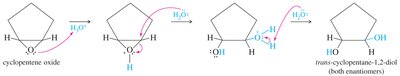
Epoxide Ring Opening
Epoxides can be opened by acid or base, typically resulting in anti addition of nucleophiles (e.g., water) to give trans-1,2-diols.
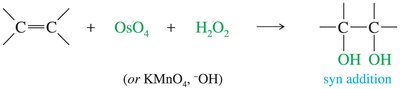
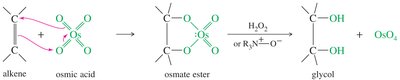
Syn Dihydroxylation
Syn dihydroxylation adds two hydroxyl groups to the same side of the alkene, forming a cis-1,2-diol.
Common reagents: osmium tetroxide (OsO4) or cold, dilute potassium permanganate (KMnO4).
Oxidative Cleavage of Alkenes
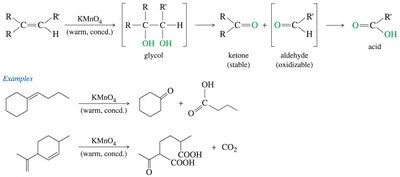
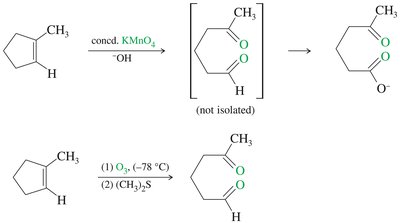
Permanganate Cleavage
Warm, concentrated KMnO4 cleaves alkenes to give ketones and/or carboxylic acids, depending on the substitution pattern.
Over-oxidation can occur, especially with aldehyde intermediates.
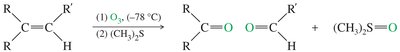
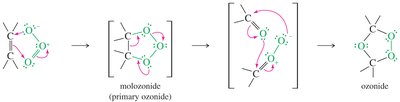
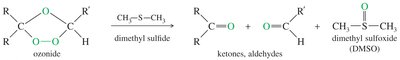
Ozonolysis
Ozone (O3) cleaves alkenes at low temperature, forming ozonides that are reduced in situ to give aldehydes and/or ketones.
Common reducing agents: zinc or dimethyl sulfide (DMS).
Ozonolysis is milder than permanganate cleavage and does not over-oxidize aldehydes.
Comparison of Permanganate Cleavage and Ozonolysis
Permanganate: Strong oxidant, can over-oxidize aldehydes to acids.
Ozonolysis: Milder, stops at aldehyde or ketone stage.
Additional Alkene Reactions
Catalytic Hydrogenation
Hydrogenation of alkenes with catalysts (e.g., Pt, Pd, or Wilkinson’s catalyst) adds H2 across the double bond in a syn fashion, converting alkenes to alkanes.
Polymerization
Alkenes can undergo cationic, radical, or anionic polymerization to form long-chain polymers.
Initiators include strong acids, BF3, or peroxides with UV light.
Summary Table: Alkene Addition Reactions
Reaction | Reagents | Regiochemistry | Stereochemistry | Product |
|---|---|---|---|---|
Oxymercuration–Demercuration | Hg(OAc)2, H2O; NaBH4 | Markovnikov | Anti addition | Alcohol |
Hydroboration–Oxidation | BH3, H2O2, NaOH | Anti-Markovnikov | Syn addition | Alcohol |
Epoxidation | Peroxyacid (e.g., mCPBA) | — | Syn addition | Epoxide |
Epoxide Opening | H2O, acid/base | — | Anti addition | Trans-1,2-diol |
Syn Dihydroxylation | OsO4, H2O2 or cold KMnO4 | — | Syn addition | Cis-1,2-diol |
Oxidative Cleavage | KMnO4 (hot, conc.) or O3, (CH3)2S | — | — | Ketones, aldehydes, acids |
