
Textbook Question
Answer the following questions for autosomal conditions such as PKU.
If both parents are heterozygous carriers of a mutant allele, what is the chance that their first child will be homozygous recessive for the mutation?
394
views

 Verified step by step guidance
Verified step by step guidance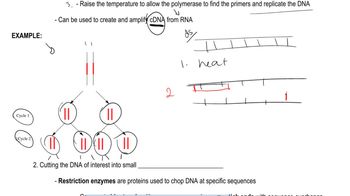
 07:39
07:39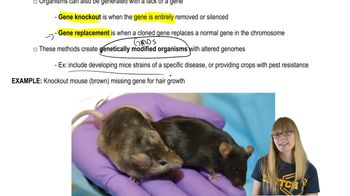 09:12
09:12Answer the following questions for autosomal conditions such as PKU.
If both parents are heterozygous carriers of a mutant allele, what is the chance that their first child will be homozygous recessive for the mutation?
Answer the following questions for autosomal conditions such as PKU.
Parents who are each heterozygous carriers for a recessive mutant allele have a child who does not have the condition. What is the chance this child is a heterozygous carrier of the condition?
Answer the following questions for autosomal conditions such as PKU.
If the first child of parents who are both heterozygous carriers of a recessive mutant allele is homozygous recessive, what is the chance the second child of the couple will be homozygous recessive? What is the chance the second child will be a heterozygous carrier of the recessive mutation?