Textbook Question
What is a reference genome? How can it be used to survey genetic variation within a species?
611
views

 Verified step by step guidance
Verified step by step guidance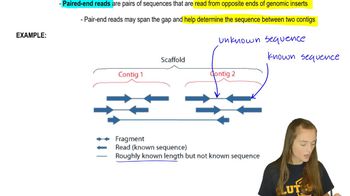
 08:41 08:41
08:41 08:41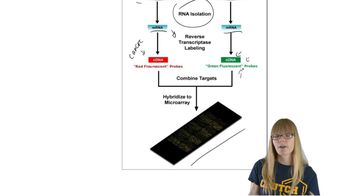 08:26
08:26What is a reference genome? How can it be used to survey genetic variation within a species?
The two-hybrid method facilitates the discovery of protein–protein interactions. How does this technique work? Can you think of reasons for obtaining a false-positive result, that is, where the proteins encoded by two clones interact in the two-hybrid system but do not interact in the organism in which they naturally occur? Can you think of reasons you might obtain a false-negative result, in which the two proteins interact in vivo but fail to interact in the two-hybrid system?
Go to http://blast.ncbi.nlm.nih.gov/Blast.cgi and follow the links to nucleotide BLAST. Type in the sequence below; it is broken up into codons to make it easier to copy.
5' ATG TTC GTC AAT CAG CAC CTT TGT GGT TCT CAC CTC GTT GAA GCTTTG TAC CTT GTT TGC GGT GAA CGT GGT TTC TTC TAC ACT CCT AAG ACT TAA 3'
As you will note on the BLAST page, there are several options for tailoring your query to obtain the most relevant information. Some are related to which sequences to search in the database. For example, the search can be limited taxonomically (e.g., restricted to mammals) or by the type of sequences in the database (e.g., cDNA or genomic). For our search, we will use the broadest database, the 'Nucleotide collection (nr/nt).' This is the nonredundant (nr) database of all nucleotide data (nt) in GenBank and can be selected in the 'Database' dialogue box. Other parameters can also be adjusted to make the search more or less sensitive to mismatches or gaps. For our purposes, we will use the default setting, which is automatically presented. Press 'BLAST' to search. What can you say about the DNA sequence?
Consider the phylogenetic trees below pertaining to three related species (A, B, and C) that share a common ancestor (last common ancestor, or LCA). The lineage leading to species A diverges before the divergence of species B and C.
For gene X, no gene duplications have occurred in any lineage, and each gene X is derived from the ancestral gene X via speciation events. Are genes AX, BX, and CX orthologous, paralogous, or homologous?
Consider the phylogenetic trees below pertaining to three related species (A, B, and C) that share a common ancestor (last common ancestor, or LCA). The lineage leading to species A diverges before the divergence of species B and C.
For gene Y, a gene duplication occurred in the lineage leading to A after it diverged from that, leading to B and C. Are genes AY1 and AY2 orthologous or paralogous? Are genes AY1 and BY orthologous or paralogous? Are genes BY and CY orthologous or paralogous?
Consider the phylogenetic trees below pertaining to three related species (A, B, and C) that share a common ancestor (last common ancestor, or LCA). The lineage leading to species A diverges before the divergence of species B and C.
For gene Z, gene duplications have occurred in all species. Define orthology and paralogy relationships for the different Z genes.