


 09:09
09:09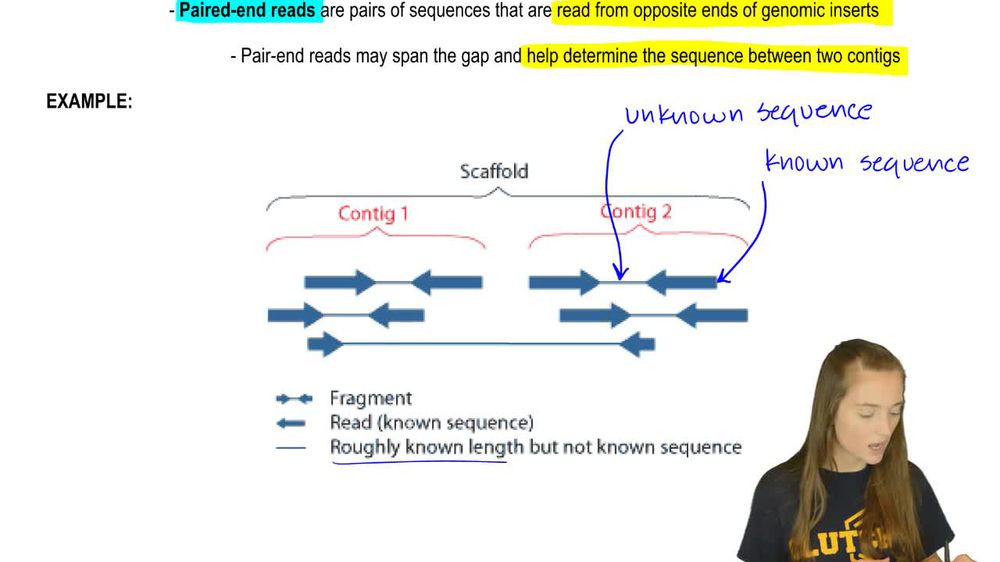

Textbook Question
You have just obtained 100 kb of genomic sequence from an as-yet-unsequenced mammalian genome. What are three methods you might use to identify potential genes in the 100 kb? What are the advantages and limitations of each method?
617
views