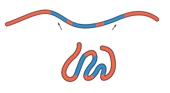
Protein folding is a complex process influenced by various factors, primarily non-covalent interactions, with the hydrophobic effect playing a crucial role. The final conformation of a protein is determined by its primary structure, which consists of the sequence of amino acids. During folding, non-polar hydrophobic amino acids tend to cluster in the protein's interior, while polar hydrophilic amino acids are positioned on the exterior, interacting with the surrounding aqueous environment.
In an unfolded polypeptide chain, hydrophobic amino acids are represented by red spheres, while hydrophilic amino acids are shown as blue spheres. The hydrophobic effect drives the aggregation of non-polar residues away from water, leading to their placement in the protein's core. Conversely, hydrophilic residues prefer to remain on the surface, where they can interact with water molecules.
Understanding the distribution of the 20 amino acids is essential for predicting their locations post-folding. Neutral amino acids are categorized into non-polar and polar groups, with aromatic amino acids like phenylalanine, tyrosine, and tryptophan distributed among these categories. A helpful mnemonic for non-polar amino acids is "GAV LIMP," which can be expanded to "GAV LIMP way fast" to include tryptophan and phenylalanine, emphasizing their hydrophobic nature.
Charged amino acids, which have full charges at physiological pH, are highly hydrophilic and are best remembered with the mnemonic "dragons eat knights riding horses." These residues are predominantly found on the protein's surface, facilitating interactions with water. Polar amino acids, while neutral, still exhibit some hydrophilic properties and can be found both inside and outside the protein, often distributed evenly. The mnemonic "Santa's team crafts new quilts" aids in recalling the polar amino acids, with tyrosine included by modifying the phrase slightly.
In summary, the arrangement of amino acids during protein folding is a critical aspect of protein structure, influencing functionality and interactions. Understanding these principles is foundational for further exploration of protein behavior and characteristics.